口罩在韩国,美国,欧盟等国家的进口要求
截止目前有据可查的全球新冠病例达到了60万之多,全球所有国家都缺乏防疫物资,口罩,防护服,呼吸机等等。而相对来说,中国已经复工复产,口罩,防护服等许多的防疫物产产品已经在生产线上,或者开始售卖。因为前些时间大部分防疫物资都要优先满足国内的需求,当目前国内形势变好的情况下,防疫物资在国内的需求慢慢在减少,因此更多的企业已经将市场瞄向海外了,那么如何将防疫物资,特别是口罩顺利出口到国外呢?各个国家又有什么不同?
口罩按其预期用途,可以分为个人防护口罩和医用口罩两大类别,在各国依据不同的法规在管理。
个 人 防 护 口 罩
市场最常见的个人防护口罩有N95/ N99、KN95、FFP2、FFP3这几种,这些是按不同国家标准而定义的口罩,主要包括:
美国 NIOSH
NIOSH: National Institute for Occupational Safety and Health美国国家职业安全卫生研究所。
根据联邦法规42 CFR Part 84,NIOSH将其认证的防颗粒物口罩分为9类,具体的测试则由NIOSH下属的NPPTL (National Personal Protective Technology Laboratory)实验室操作。
根据口罩中间滤网过滤特性分为下列三种:
N系列:N代表Not resistant to oil,可用来防护非油性悬浮微粒。
R系列:R代表Resistant to oil,可用来防护非油性及含油性悬浮微粒。
P系列:P代表oil Proof,可用来防护非油性及含油性悬浮微粒。
按滤网材质的最低过滤效率,又可将口罩分为下列三种等级:
95等级:表示最低过滤效率为95% ;
99等级:表示最低过滤效率为99% ;
100等级:表示最低过滤效率为99.97% ;
组合起来就为N100, N99, N95, R100, R99, R95, P100, P99, P95.共9种。
上述9种口罩需满足美国联邦法规42 CFR Part 84的要求,主要测试指标包括呼气阻力测试(Exhalation Resistance Test)、呼气阀泄漏测试(Exhalation Valve Leakage Test)、吸气阻力测试(Inhalation Resistance Test)、过滤效率测试(Sodium Chloride Test)。
认证的申请:需按照NIOSH的指南实施,企业需寄送样品至NIOSH实验室实施测试,同时提交技术性资料(包括质量体系部分资料)至NIOSH文审,只有文审和测试都通过,NIOSH才核发批文。
欧洲 PPE
口罩需依据欧洲个人防护法规PPE (Personal Protective Equipment) Regulation (EU) 2016/425实施申请,口罩主要分类FFP2、FFP3。
企业需选择有PPE法规授权的公告机构(Notified body)实施申请,NB机构需审核企业质量管理体系和CE技术文档。审核通过后可获得PPE法规的CE证书。
中国 KN系列
按国标GB2626-2006《呼吸防护用品自吸过滤式防颗粒物呼吸器》的要求,将口罩分为三个等级:KN90、KN95、KN100。过滤效率分别为:90%、95%、99.97% ;企业需测试通过后才可声称满足KN系列要求;
中国 NMPA:依据NMPA 2017年8月发布的《医疗器械分类目录》,产品分类管理类别为:II类医疗器械 。中国对医用的口罩分类较细,分为三个类别:医用防护口罩、医用外科口罩和一次性使用医用口罩。
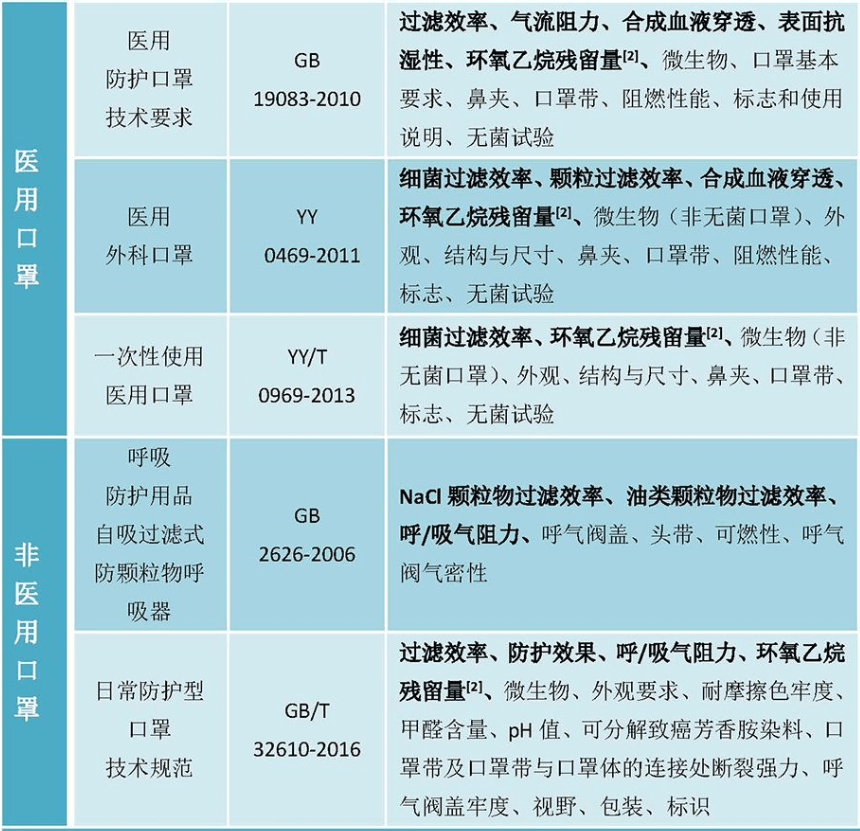
按技术要求来看,医用防护口罩要求最高,其次医用外科口罩,最后为一次性使用医用口罩。
步骤流程
1,依据产品《技术要求》委托CNAS授权的各省医疗器械检测所进行产品的测试;
2,准备注册文件,结合测试报告等向省局药监局申报;
3,注册文件齐全后向省药监局申报,省药监局受理;
4,药监局审理注册文件的同步,派出审核官对制造商进行现场GMP体系考核;
5,注册文件通过审核,现场通过审核,颁发产品注册证书;
6,制造商收到注册证书后准备生产许可证申请资料,一般向市市场监督管理局申请颁发生产许可证书;
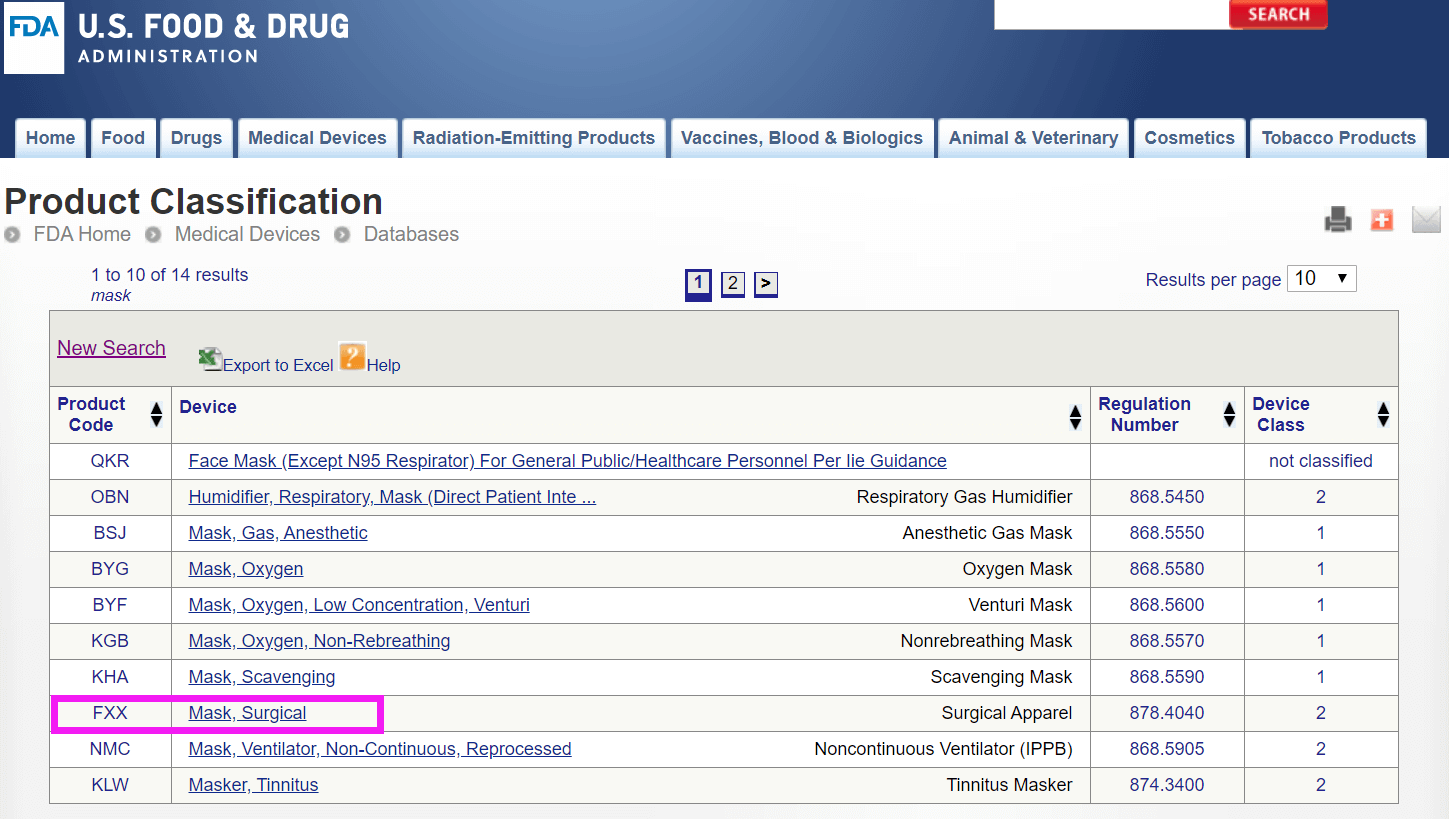
美国 FDA
依据美国FDA分类: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
依据关键词" Mask ",进行搜索: 选择最合适的代码:FXX,依据FDA指南编写510k报告,提交美国FDA审批。
关键技术点
1)产品的性能测试包括:
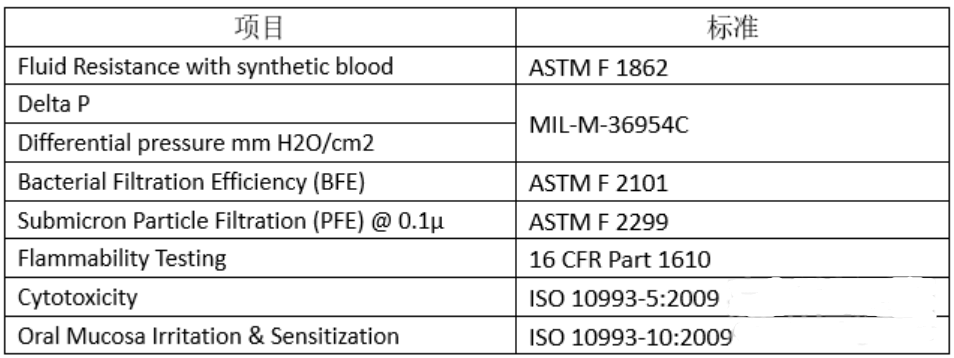
2)该产品不需要提交灭菌确认报告等。
3)费用及周期:美国FDA 510k审核费,2020年正常官方费用:$5,236,小规模企业可申请优惠:$2,899;正常周期,启动项目至审批10个月左右。
欧盟 CE
依据Regulation(EU)2017/745 on medical devices即MDR;依据Annex VIII分类规则10,Class Is,需要公告参与审核体系ISO13485:2016及CE技术文件。
费用及周期:
由于欧盟采用的是委托具备资质的第三方公告机构监管方式,审核并颁发证书,相关费用以NB机构报出的为准,鉴于目前已获得资质的10家机构还未全面进行业务受理,周期不得而知,依据MDD的经验,预计12-15个月左右。
医疗口罩材料:性能专标EN14683-2014
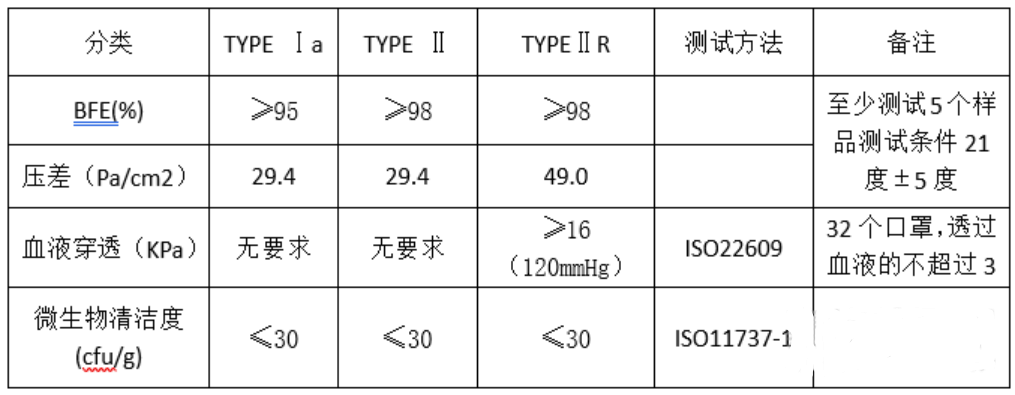
鉴于当前疫情下,建议选择一类非无菌Class I方式进行编写MDR技术文档,自我声明,选择欧代,进行欧盟注册,加贴CE标识,产品出口欧盟。
韩国 KFDA
韩国医疗器械准入的法规门槛,基本分类为I、II、III、IV类,持证为韩国公司(License holder),且韩代职责较重,如:快速通关、快速到门的服务、仓库管理,包括韩国保址部所有监管事宜而且在海关业务的应急时间等全链条内容,接受KGMP定期评审等。
KFDA的等级标准:
●等级I:非接触人体的或无潜在危险性的产品.
●等级II:对人体具有一定的危险性但对人体生命 的危险及造成的影响和危险性比较低的产品.
●等级III:一定时间内插入人体使用或潜在的危险性比较高的产品.
●等级IV:永久的移植到人体内或直接接触到心脏、中枢神经神经等而使用的产品
韩国注册相关内容:
1)申请KGMP证书和接受现场审核;
2)对于II类产品一般是委托授权的第三方审核员完成审核(但笔者前期服务的案例中,也都是韩国保址部官员),若为III IV类产品则由韩国保址部(相当于中国药监局)自行审核,并获得KGMP证书;
3)寄送样品到韩国MFDS授权的实验室进行韩标的测试;
4)由韩代向MFDS提交技术文件(包括TCF、检测报告、KGMP证书等),同时还需要向韩国保址部缴纳申请费,后续由该机构进行注册文件的审核,最终获得批准,后续方可入市。
5)医用外科口罩属于韩国II类产品,预计周期6-9个月。
Say Something!